1.什么是LAMMPS
LAMMPS:Large-scale Atomic/Molecular Massively Parallel Simulator
是Steve Plimpton为主要作者开发一款开源的分子动力学模拟软件。自2004年发布以来,LAMMPS已被下载数十万次,并已发展成为全球领先的基于粒子的材料建模代码。
LAMMPS可以模拟固态(金属,非金属,半导体等),液态(水,溶液),软物质(高分子,DNA,蛋白质),粗粒化物质。甚至可以这样说,只要能用分子动力学模拟的东西,LAMMPS就能搞定。事实上,LAMMPS作为一款并行粒子模拟器,可以在原子,介观和连续介质尺度上开展高效模拟。在计算能力和效率方面,LAMMPS可以支持十亿级原子规模的计算,可同时调用上万个核和数百个GPU进行计算。
它可以在任何可以编译C++和支持MPI数据通讯库的并行计算机上工作
为什么分子动力学模拟如此重要?
这要从我们对这个世界的认知开始讲起。20世纪著名物理学家理查德·费曼在其传世物理教材《费曼物理讲义》中提出这样一个有意思的问题:“假如由于某种大灾难,所有的科学知识都丢失了,只有一句话传给下一代,那么怎样才能用最少的词汇来表达最多的信息呢?”费曼认为这句话应该是原子假设:“所有物体都是用原子构成的——这些原子是一些小小的粒子,它们一直不停地运动着,当彼此略微离开地时互相吸引,当彼此过于靠近时又相互排斥。”这句话之所以重要是因为基于这个原子假设我们能解释巨量自然界中的现象。比如,气体压强来自于大量气体分子对壁面撞击;水蒸发吸热来自于水分子从体相水中逃逸至外界环境而带走能量所致......
费曼把原子假设放在如此高度的原因总结起来就是——掌握组成物体内部原子的行为就可解释和理解物理的性质和行为特性。拉普拉斯也曾有过类似的表述,即物理学的四大神兽之一——拉普拉斯兽。拉普拉斯兽是这样一只神奇的动物,它能掌握世界上所有原子的位置和动量并能加以分析(它能够同时观察到宇宙中的每一个粒子,并将它们的状态以数据的形式表示出来),因此世界上发生的一切及其背后的原因都一览无余的呈现在它眼前。这些观点基于经典力学对世界的认识。这一认识已经足够我们解释生活和工程中遇到的大多数现象
lammps的发展现况
LAMMPS性能最好,接口齐全,有GPU等各种加速包,力场最全,还是开源的,极易扩展;其次,手册完整,邮件组活跃(24小时有18个小时,发出的问题可以在15分钟内得到核心维护人员的回复);再者,处于高频的更新,bug fix半个月一次,版本更新三个月一次,向下兼容
2.HPC与LAMMPS的关系
说道这个问题,我们可以从lammps的起源说起:lammps有两位最主要的开发者——
Axel Kohlmeyer: Steve Plimpton:
Steve Plimpton:
前者是美国天普大学的教授(现阶段lammps的主要维护者),计算分子科学研究所副所长和高性能计算 (HPC) 团队的负责人。他还是意大利的里雅斯特国际理论物理中心的外部助理,在那里他与 ICTP HPC 工作人员合作开展 HPC 相关活动,并在由 ICTP 和 SISSA 在的里雅斯特举办的国际 HPC 硕士课程中任教。总被引次数2395,H因子21
后者更是重量级(lammps、SPPARKS、SPARTA之父),他研究主要集中在各种基于粒子的 HPC 应用的方法和并行算法上,他还致力于在HPC平台上开发基于MPI的大数据处理工具。总被引次数达到55486次,从2017年以来的引用达到32084,H因子60。LAMMPS的主要使用算法论文单篇引用达到37049。于2017年获得Fernbach奖,Fernbach奖于 1992 年设立,以纪念 HPC 先驱 Sidney Fernbach,旨在表彰产生出色 HPC 结果的创新方法,是 IEEE 的最高奖项之一。
从这两个主要开发者的履历就可以看出来,HPC和lammps有着非常精密的联系,甚至可以说lammps就是基于hpc在运转的软件(当然一些简单的例子并不需要hpc,家用电脑就可以运行);
分子动力学实际上就是一只拉普拉斯兽:
分子动力学的基本任务就是获取物体在任意时刻组成原子的所有位置和动量然后利用统计力学知识理解物体的性质和行为。如果这样还不能说服你分子动力学的实力。就讲一个事实——2013年诺贝尔化学奖颁给了创立跨尺度分子模拟的三位科学家。通过分子动力学模拟我们可以深刻理解我们所研究的对象。目前各种硬件超强的计算能力为分子动力学创建了一个大有可为的时代!!没有HPC的强大算力,分子动力学的模拟就是一纸空谈;这也是为什么HPC被称为科学的第三支柱。

3.Lammps安装
3.0 lammps文件结构
LAMMPS执行程序(lmp_mpi)
脚本文件(in文件)
势函数文件(eam/alloyetc.)
Data文件(体系的初始构型)
MPI并行文件(多核并行运算)
默认情况下,LAMMPS通过读取脚本文件中的命令运行。因此仅仅是运行可执行文件(lmp_mpi)它会一直在等待,直到有命令从键盘输入。通常,对于并行的环境应该使用这样的命令输入一个脚本:
lmp_mpi –in in.file
3.1 Windows安装
首先先去官网:https://www.lammps.org/
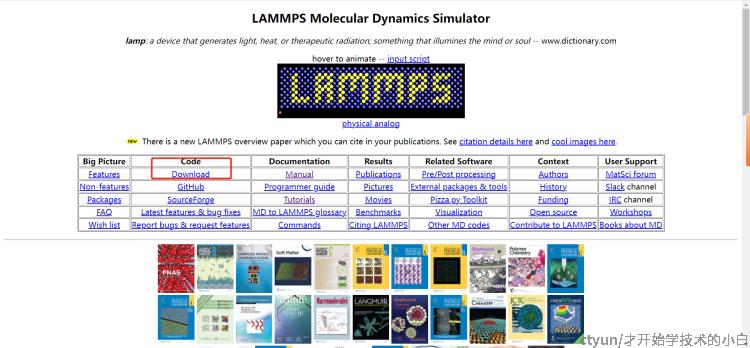
选择pre-build windows
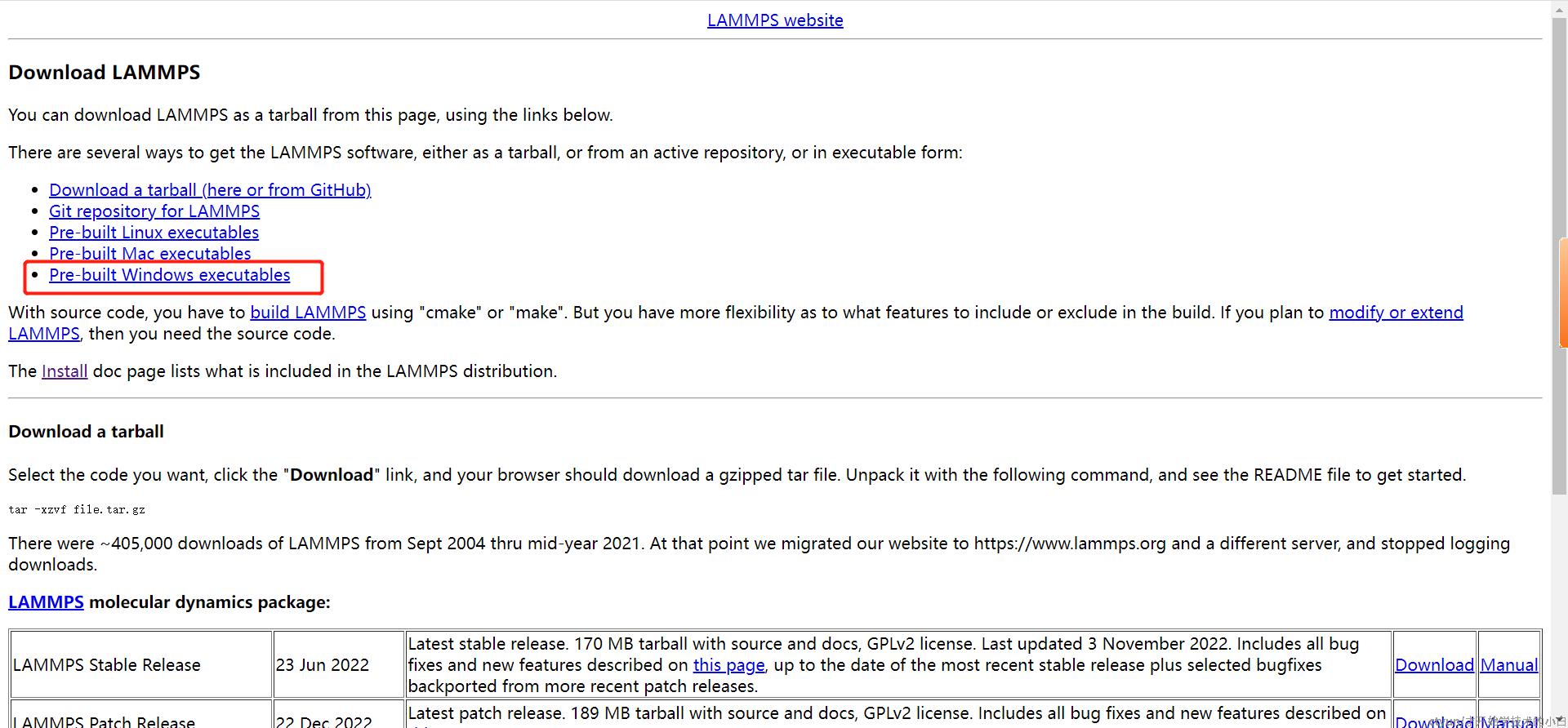
会跳转到该页面,进入这个网址即可:
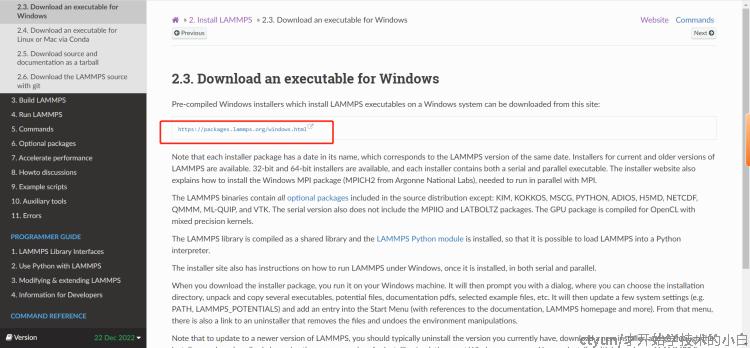
找到windows,进入64位安装:
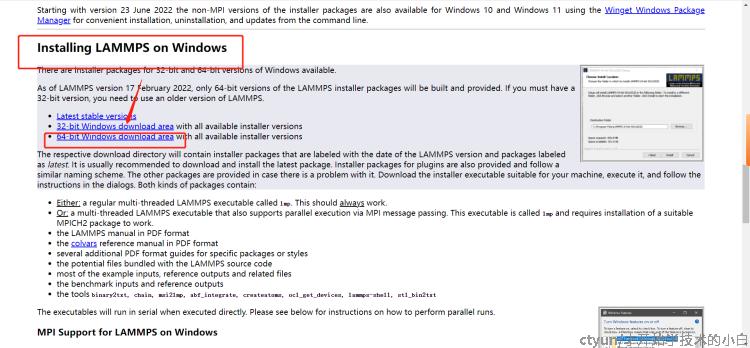
选择最新的并行版本(带MPI的)安装包
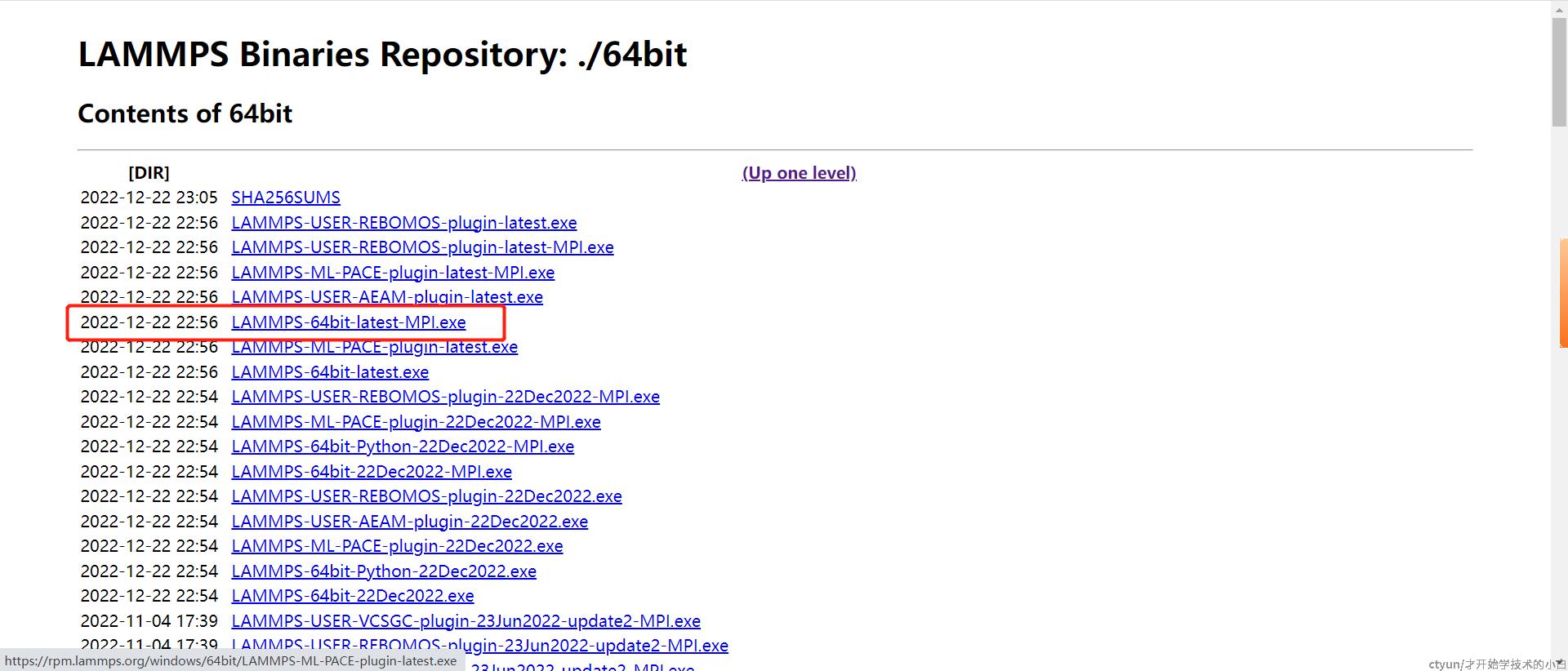
回到上一个界面,下载64位的mpi工具:
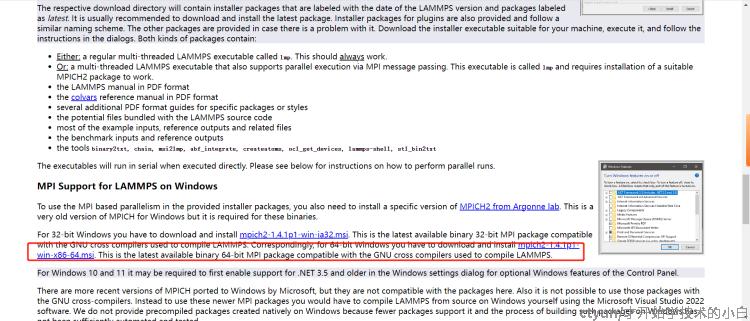
右键管理员运行lammps的安装程序,默认安装即可
接下来右键管理员身份安装mpi,同样执行默认安装:
接来下进入lammps的安装目录,我们这里是在:
C:\Users\WY\AppData\Local\LAMMPS 64-bit 22Dec2022-MPI
C:\Program Files\MPICH2\
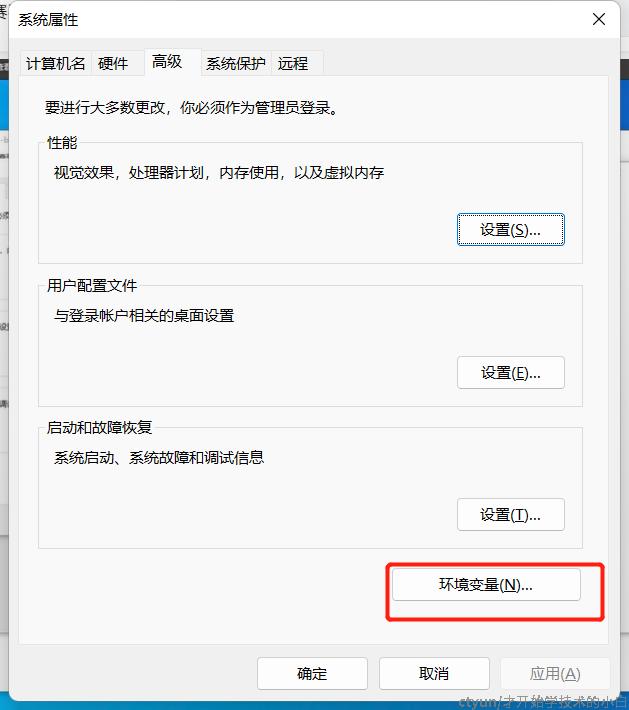
开始编辑环境变量,先进入系统属性-环境变量
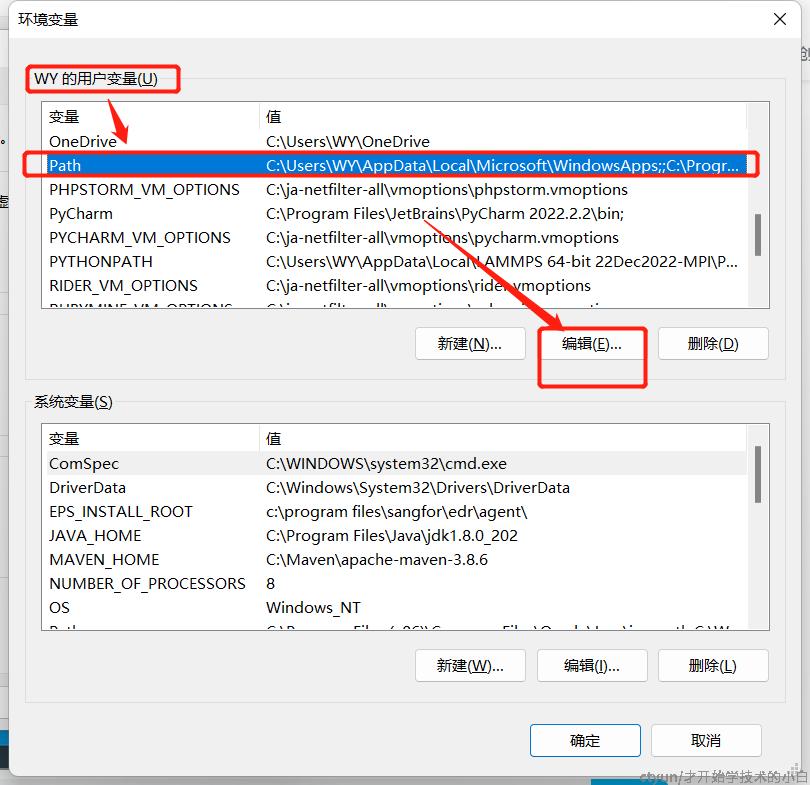
在用户变量下的path更改:
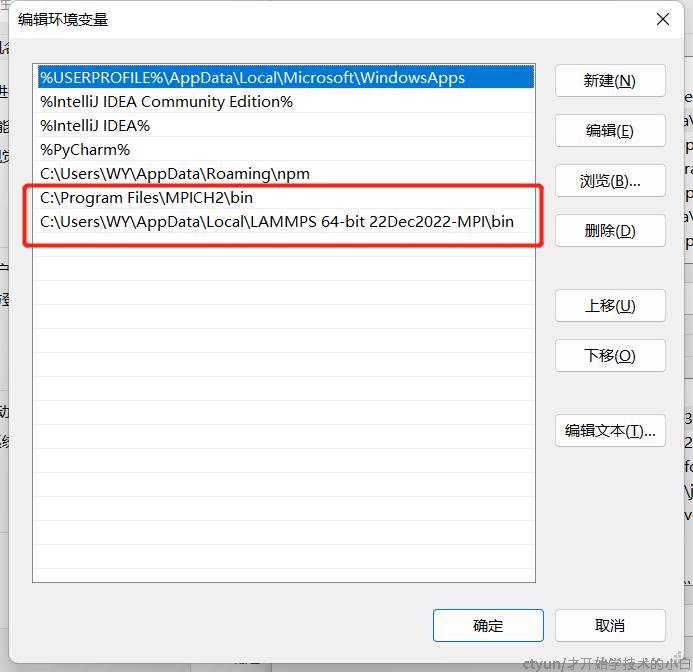
这里进去可以看到已经有lammps和mpi的目录,但是如果你在安装mpi的时候更改了默认路径,则这个并不是安装路径下的,还是需要手动配置一下你更改的路径
打开命令行,一定要以管理员身份运行,进入到lammps的安装路径,执行以下操作:
#进入安装路径
cd C:\Users\WY\AppData\Local\LAMMPS 64-bit 22Dec2022-MPI
#进入bin
cd bin
#安装MPICH2 Process Manager, Argonne National Lab
smpd -install -phrase behappy
#移除以前安装过的信息,如果报错也没事,可能是之前没装过,是空信息
mpiexec -remove
#注册信息
mpiexec -register这里会提示输入管理员名称,可以在控制面板-用户账户中查看(如下图)我们这里是WY
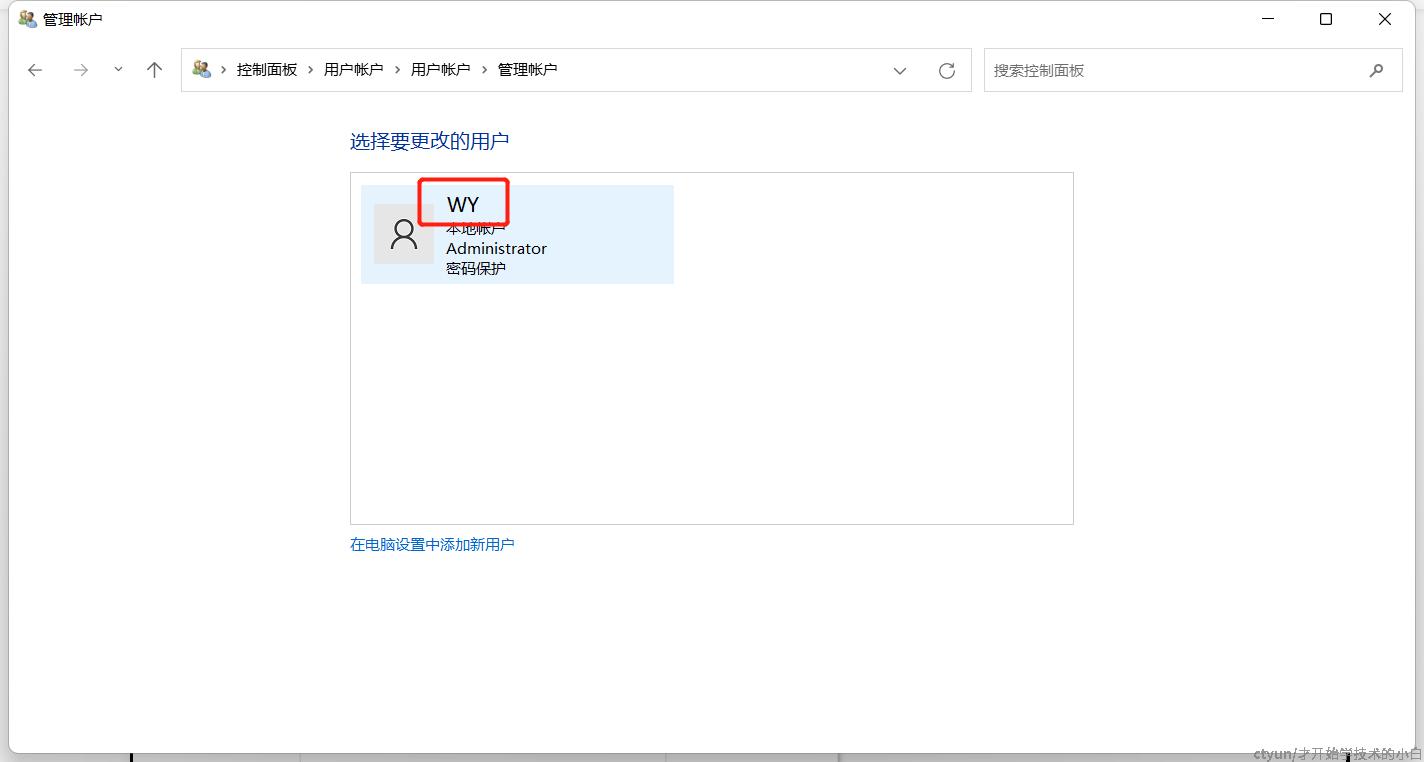
输入WY,再输入开机密码(注意不是pin码),出现以下界面则成功了(也可以使用mpiexec -validate看下返回是不是SUCCESS,或者用lmp命令查看安装信息)

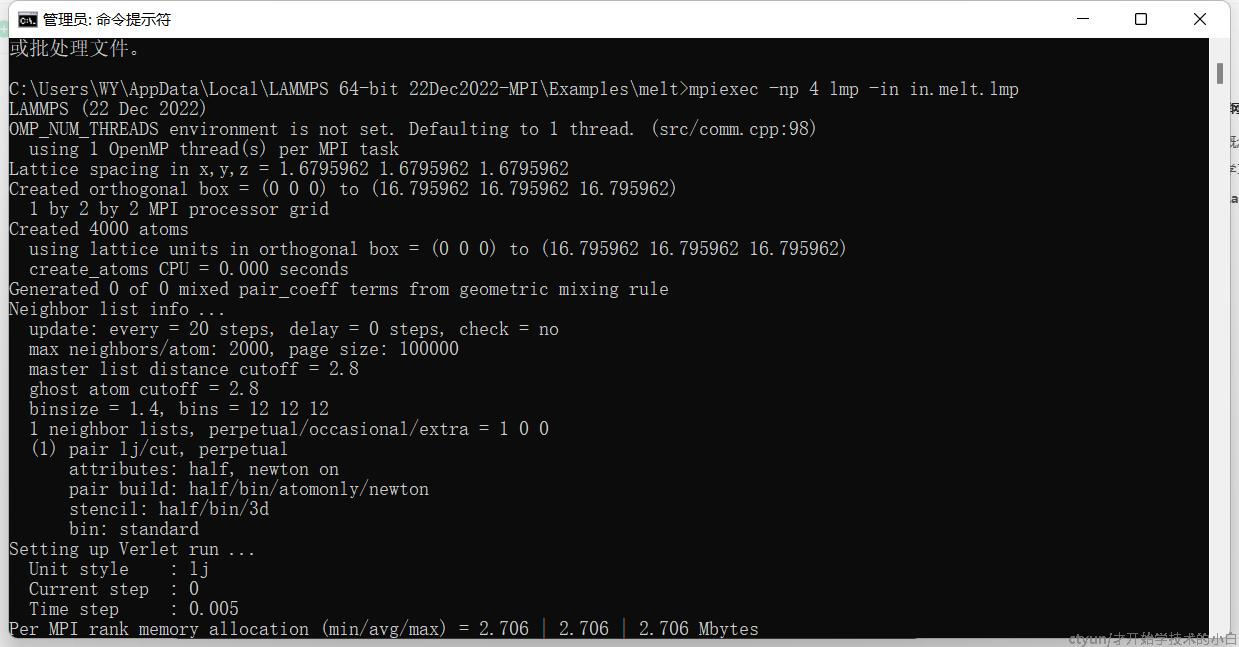
可以使用命令行来执行自带的example,比如melt这个例子,我们先cd到这个目录
cd C:\Users\WY\AppData\Local\LAMMPS 64-bit 22Dec2022-MPI\Examples\melt
#用四个核心并行执行
mpiexec -np 4 lmp -in in.melt.lmp结果如下:非常顺利
3.2 CentOS安装
绝大部分使用lammps的客户,都是在服务器上运行,相对windows,centOS是服务器端的绝对主力系统,因此了解CenOS下如何集成lammps是对我们hpc产品发展非常有利的,可以提前下载/更新对应的依赖:
● gcc
● gcc-c++
● fftw
● fftw-devel
● openmpi
● openmpi-devel
# 1.下载相关软件包和依赖软件(可以加-b选项后台安装)
# lammps
wget https://lammps.sandia.gov/tars/lammps-stable.tar.gz
# mpich,用来实现并行计算
wget https://www.mpich.org/static/downloads/3.2.1/mpich-3.2.1.tar.gz
# fftw,快速计算离散傅里叶变换的标准C语言程序集
wget https://www.fftw.org/fftw-3.3.8.tar.gz
# 这里我们将三个包都放在lammps文件夹下
mkdir lammps
mv lammps-stable.tar.gz /root/lammps
mv mpich-3.2.1.tar.gz /root/lammps
mv fftw-3.3.8.tar.gz /root/lammps
# 2.安装fftw
tar -zxvf fftw-3.3.8.tar.gz
cd fftw-3.3.8/
./configure
make
make install
# 3.安装mpich
cd ..
tar -zxvf mpich-3.2.1.tar.gz
cd mpich-3.2.1/
./configure
# 配置可能会报错,需要fortran 77,安装一个即可
yum install gcc-gfortran
make
make install
# 安装完可以测试下:输入mpicx,按table键看能否自动出现mpicxx这个命令
# 4.安装lammps
cd ..
tar -zxvf lammps-stable.tar.gz
cd lammps-23Jun2022/src/
# 编译lammps时安装所有包
make yes-all
# 取消需要外链的包
make no-lib
#编译
make mpi
# 5.配置环境变量
cd
vi .bashrc
# 添加如下行
export PATH=~/lammps-23Jun2022/src:$PATH
source .bashrc
# 6.测试lammps
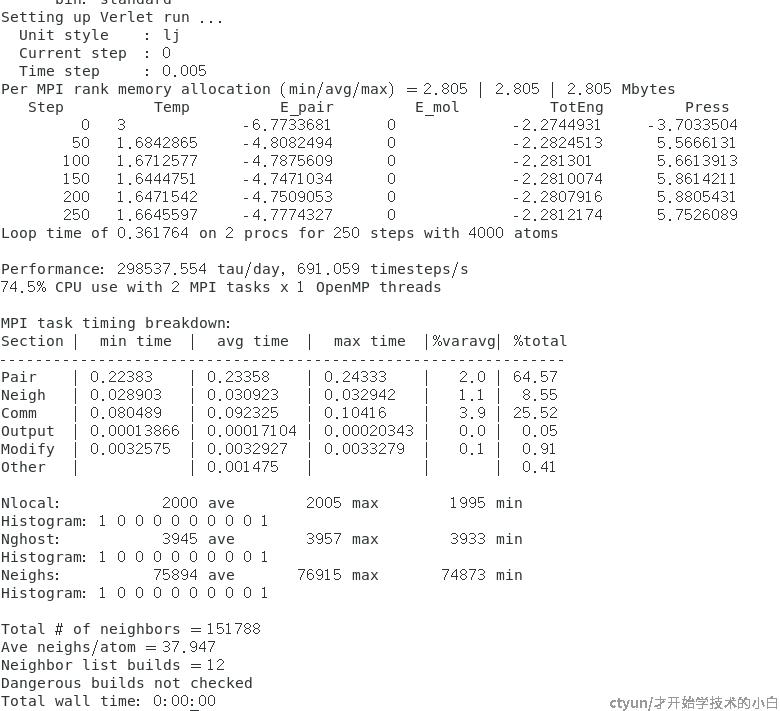
# 进入安装目录下的example,我们拿melt来试试
/examples/melt
mpirun -np 2 lmp -in in.melt最后结果如图:
下一期介绍lammps计算出来的数据的可视化过程,以及如何用超算集群去调用lammps