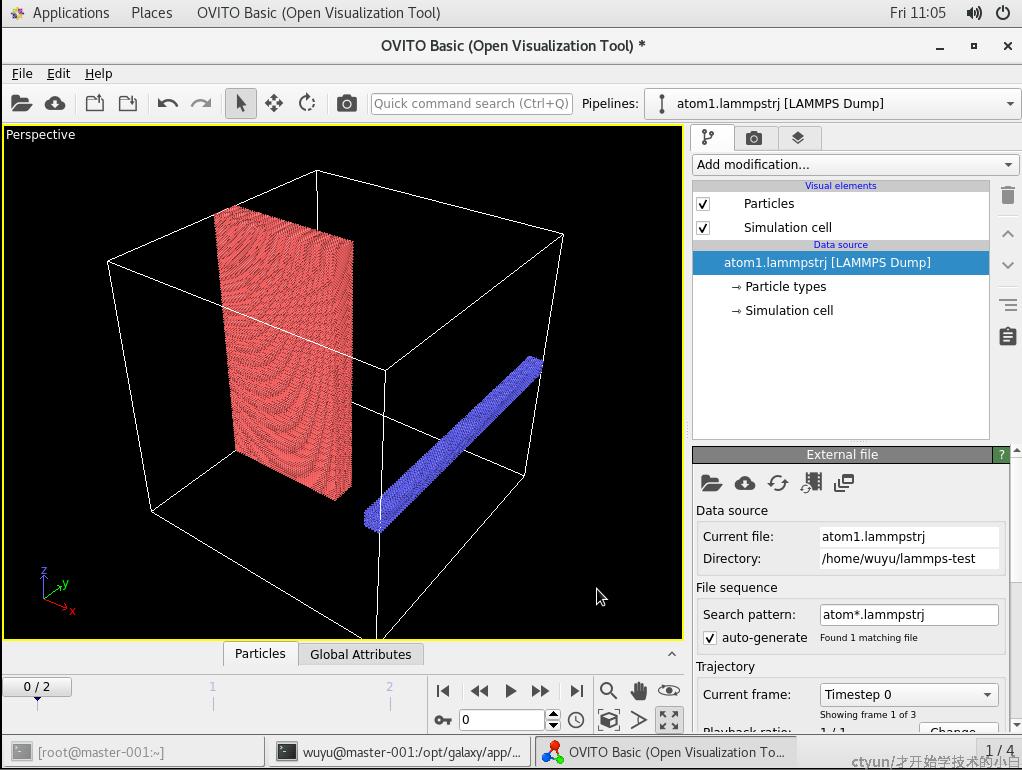
1.lammps的数据可视化
通常我们使用ovito/vmd来进行数据可视化,这里着重讲ovito,一方面是因为它比vmd好用(虽然不能建模),另一方面也是因为vmd的安装实在是太简单了
1.1 Windows安装ovito
还是有不少演示机器会希望直接使用Windows来看数据,所以ovito的exe下载量还是蛮高的
1.2 CentOS安装ovito
还是一样进入官网下载linux的basic版本,不用解压,直接xftp之类的工具传输过去即可
# 解压
tar -zxvf ovito-basic-3.7.12-x86_64.tar.xz
# 完成后bin目录下可以直接运行
./ovito
# 为了方便调用,我们将其设置为环境变量,由于是root安装的我们这里是:/usr/local/bin
vi .bashrc
# 加入以下行
export PATH=/usr/local/bin:$PATH
# 执行后,直接ovvito即可使用
source .bashrc1.3 vmd的安装
步骤都跟ovito一样,这几就不赘述了,但是要注意vmd打开是会出现三个窗口的,这个是正常现象
2.使用slurm运行Lammps
上一篇文章介绍了很多hpc和slurm的关系,但是在示例部分我们实际上是直接使用我们的终端(Windows/linux)来运行的,换而言之如果碰到比较复杂的模型我们应该怎么办呢?总不能给hpc装一个Windows系统吧,于是我们就需要使用hpc提交作业的脚本来提交in文件
具体的集群搭建可以参考这篇文章:https://www.ctyun.cn/developer/article/363542369067077
搭建好了集群之后,我们可以使用sbatch的方法来提交作业,sbatch一些基本的命令如下:
-o job.%j.out # 脚本执行的输出将被保存在当job.%j.out文件下,%j表示作业号;
-p C032M0128G # 作业提交的指定分区为C032M0128G;
--qos=low # 指定作业的QOS为low;
-J myFirstJob # 作业在调度系统中的作业名为myFirstJob;
--nodes=1 # 申请节点数为1,如果作业不能跨节点(MPI)运行, 申请的节点数应不超过1;
--ntasks-per-node=1 # 每个节点上运行一个任务,默认一情况下也可理解为每个节点使用一个核心,如果程序不支持多线程(如openmp),这个数不应该超过1;这里我们提交的测试脚本为:
#!/bin/bash
#SBATCH --job-name=lammps-test
#SBATCH --output=lammps-test.out
#SBATCH --partition=p1
mpirun -np 1 lmp -in in.melt
mpirun -np 1 lmp -in in.comb.Cu这样就实现了同时提交两个in文件(提交命令为:sbatch lammps-test.sh),需要注意的有以下几点:
1.需要用普通用户去创建运行文件的目录,因为lammps运行的时候需要输出日志,没创建文件的权限会报错
2.不推荐使用root用户运行,即使加了--allow-run-as-root,相关的mpi依然有可能报错
3.有可能需要用lmp_mpi而不是lmp,看lammps安装的具体情况,我们的示例中为lmp
4.需要使用 1 作为slot,slurm会自动分配多个节点来计算
5.如果缺少势文件,可以从window系统里面传输到运行的目录下,或者在lammps目录下新增一个+改变in问阿金调用势文件的路径
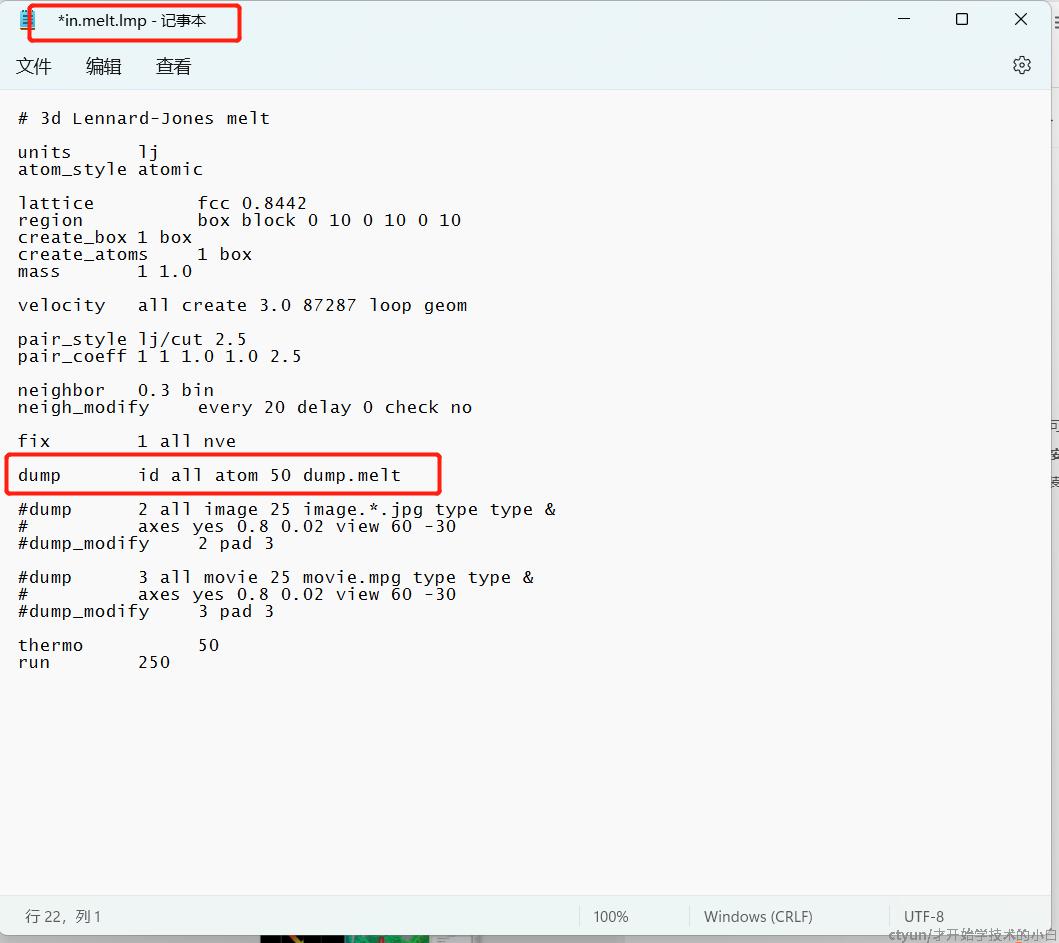
结果如下(注释掉in文件的dump就可以输出轨迹文件,进而在ovito里面观看):
LAMMPS (23 Jun 2022 - Update 2)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (src/comm.cpp:98)
using 1 OpenMP thread(s) per MPI task
Lattice spacing in x,y,z = 1.6795962 1.6795962 1.6795962
Created orthogonal box = (0 0 0) to (16.795962 16.795962 16.795962)
1 by 1 by 1 MPI processor grid
Created 4000 atoms
using lattice units in orthogonal box = (0 0 0) to (16.795962 16.795962 16.795962)
create_atoms CPU = 0.000 seconds
Generated 0 of 0 mixed pair_coeff terms from geometric mixing rule
Neighbor list info ...
update every 20 steps, delay 0 steps, check no
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 2.8
ghost atom cutoff = 2.8
binsize = 1.4, bins = 12 12 12
1 neighbor lists, perpetual/occasional/extra = 1 0 0
(1) pair lj/cut, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d
bin: standard
Setting up Verlet run ...
Unit style : lj
Current step : 0
Time step : 0.005
Per MPI rank memory allocation (min/avg/max) = 3.222 | 3.222 | 3.222 Mbytes
Step Temp E_pair E_mol TotEng Press
0 3 -6.7733681 0 -2.2744931 -3.7033504
50 1.6842865 -4.8082494 0 -2.2824513 5.5666131
100 1.6712577 -4.7875609 0 -2.281301 5.6613913
150 1.6444751 -4.7471034 0 -2.2810074 5.8614211
200 1.6471542 -4.7509053 0 -2.2807916 5.8805431
250 1.6645597 -4.7774327 0 -2.2812174 5.7526089
Loop time of 0.362727 on 1 procs for 250 steps with 4000 atoms
Performance: 297744.252 tau/day, 689.223 timesteps/s
98.9% CPU use with 1 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 0.30078 | 0.30078 | 0.30078 | 0.0 | 82.92
Neigh | 0.050034 | 0.050034 | 0.050034 | 0.0 | 13.79
Comm | 0.0051255 | 0.0051255 | 0.0051255 | 0.0 | 1.41
Output | 0.00021937 | 0.00021937 | 0.00021937 | 0.0 | 0.06
Modify | 0.005321 | 0.005321 | 0.005321 | 0.0 | 1.47
Other | | 0.001243 | | | 0.34
Nlocal: 4000 ave 4000 max 4000 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5506 ave 5506 max 5506 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 151788 ave 151788 max 151788 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 151788
Ave neighs/atom = 37.947
Neighbor list builds = 12
Dangerous builds not checked
Total wall time: 0:00:00
LAMMPS (23 Jun 2022 - Update 2)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (src/comm.cpp:98)
using 1 OpenMP thread(s) per MPI task
Lattice spacing in x,y,z = 3.615 3.615 3.615
Created orthogonal box = (0 0 0) to (14.46 14.46 14.46)
1 by 1 by 1 MPI processor grid
Created 256 atoms
using lattice units in orthogonal box = (0 0 0) to (14.46 14.46 14.46)
create_atoms CPU = 0.000 seconds
Reading comb potential file ffield.comb with DATE: 2011-02-22
Pair COMB:
generating Coulomb integral lookup table ...
element[1] = Cu, z = 0.454784
will not apply over-coordination correction ...
Neighbor list info ...
update every 1 steps, delay 1 steps, check yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 12.5
ghost atom cutoff = 12.5
binsize = 6.25, bins = 3 3 3
1 neighbor lists, perpetual/occasional/extra = 1 0 0
(1) pair comb, perpetual
attributes: full, newton on
pair build: full/bin/atomonly
stencil: full/bin/3d
bin: standard
Setting up Verlet run ...
Unit style : metal
Current step : 0
Time step : 0.0002
Per MPI rank memory allocation (min/avg/max) = 7.47 | 7.47 | 7.47 Mbytes
Step Temp TotEng PotEng E_vdwl E_coul Press Volume Lx Ly Lz Xz
0 10.1 -3.5063151 -3.5076155 -3.5076155 0 27.496055 3023.4645 14.46 14.46 14.46 0
1 10.099643 -3.5063151 -3.5076155 -3.5076155 0 27.512983 3023.4645 14.46 14.46 14.46 0
2 10.098572 -3.5063151 -3.5076153 -3.5076153 0 27.563765 3023.4645 14.46 14.46 14.46 0
3 10.096788 -3.5063151 -3.5076151 -3.5076151 0 27.64839 3023.4645 14.46 14.46 14.46 0
4 10.094291 -3.5063151 -3.5076148 -3.5076148 0 27.766843 3023.4645 14.46 14.46 14.46 0
5 10.09108 -3.5063151 -3.5076144 -3.5076144 0 27.919101 3023.4645 14.46 14.46 14.46 0
6 10.087158 -3.5063151 -3.5076139 -3.5076139 0 28.105138 3023.4645 14.46 14.46 14.46 0
7 10.082524 -3.5063151 -3.5076133 -3.5076133 0 28.324919 3023.4645 14.46 14.46 14.46 0
8 10.077179 -3.5063151 -3.5076126 -3.5076126 0 28.578403 3023.4645 14.46 14.46 14.46 0
9 10.071123 -3.5063151 -3.5076118 -3.5076118 0 28.865545 3023.4645 14.46 14.46 14.46 0
10 10.06436 -3.5063151 -3.5076109 -3.5076109 0 29.186292 3023.4645 14.46 14.46 14.46 0
Loop time of 0.100091 on 1 procs for 10 steps with 256 atoms
Performance: 1.726 ns/day, 13.902 hours/ns, 99.909 timesteps/s
98.1% CPU use with 1 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 0.099538 | 0.099538 | 0.099538 | 0.0 | 99.45
Neigh | 0 | 0 | 0 | 0.0 | 0.00
Comm | 0.00031282 | 0.00031282 | 0.00031282 | 0.0 | 0.31
Output | 0.00015368 | 0.00015368 | 0.00015368 | 0.0 | 0.15
Modify | 4.3161e-05 | 4.3161e-05 | 4.3161e-05 | 0.0 | 0.04
Other | | 4.356e-05 | | | 0.04
Nlocal: 256 ave 256 max 256 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 4375 ave 4375 max 4375 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 0 ave 0 max 0 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 172544 ave 172544 max 172544 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 172544
Ave neighs/atom = 674
Neighbor list builds = 0
Dangerous builds = 0
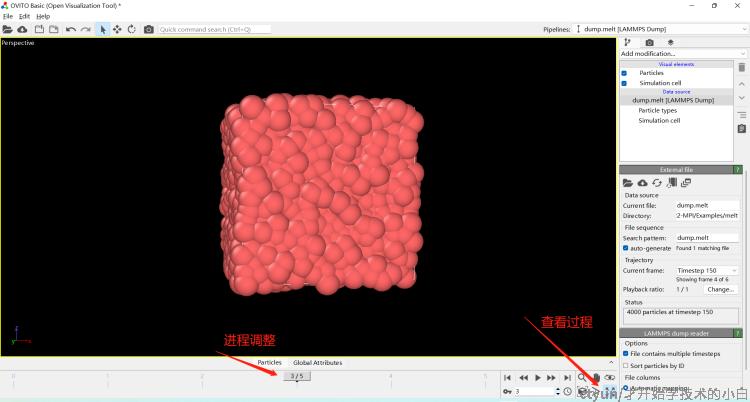
Total wall time: 0:00:00如果导入ovito,结果如图;